Neurodegeneracja związana z kinazą pantotenianu
| morbus Hallervorden-Spatz | |
| Klasyfikacje | |
| ICD-10 | G23.0 |
|---|---|
Neurodegeneracja związana z kinazą pantotenianu (także encefalopatia z odkładaniem żelaza, neurozwyrodnienie z akumulacją żelaza typu I, dawniej choroba Hallervordena-Spatza, ang. panthotenate kinase-associated neurodegeneration, PKAN, Hallervorden-Spatz disease) – genetycznie uwarunkowana choroba neurodegeneracyjna, jedna z najczęstszych chorób układu pozapiramidowego u dzieci. Chorobę opisali Hugo Spatz i Julius Hallervorden w pracy z 1922 roku.
Etiologia
Choroba jest spowodowana mutacją w genie PANK2 w locus 20 p13-p12.3. Dziedziczy się w sposób autosomalny recesywny. Występuje z częstością 1-3: 1 000 000 osób[1]. Gen PANK2 koduje kinazę pantotenianową 2, enzym biorący udział w biosyntezie koenzymu A, pantotenianu (witaminy B5) i panteteiny. Wiadomo, że w neuronach jąder podstawy chorych na HSS odkładają się duże ilości żelaza, prawdopodobnie działającego neurotoksycznie przez indukcję powstawania reaktywnych form tlenu.
Objawy i przebieg
Wyróżnia się cztery typy choroby według Seitelbergera:
- typ dziecięcy
- typ późnodziecięcy
- typ młodzieńczy
- typ dorosłych.
Pierwsze objawy najczęstszej postaci późnodziecięcej pojawiają się między 4. a 15. rokiem życia. Są to:
- otępienie
- uogólniona dystonia, zwłaszcza oromandibularna
- dysfagia
- dyzartria
- ruchy choreoatetotyczne
- spastyczność, stopa końsko-szpotawa lub wydrążona (pes excavatum)
- tylnosznurowe zaburzenia czucia
- czasem pogorszenie widzenia spowodowane barwnikowym zwyrodnieniem siatkówki), do ślepoty włącznie.
W postaci dorosłych przeważają objawy zespołu parkinsonowskiego.
Rozpoznanie
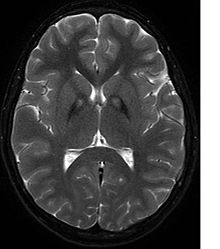
Rozpoznanie opiera się na badaniu MRI mózgu T2-zależnym, wykazujące charakterystyczne zmiany hiperintensywne w przednioprzyśrodkowej części gałki bladej i obniżenie sygnału z zewnętrznej warstwy gałki bladej i części siatkowatej istoty czarnej[2], tzw. objaw tygrysiego oka, charakterystyczny, ale nie patognomoniczny dla choroby Hallervordena-Spatza[3]. Badanie neuropatologiczne wykazuje obecność złogów żelaza i α-synukleinododatnich sferoidów w gałce bladej.
Leczenie
Nie ma jak dotąd możliwości leczenia przyczynowego choroby, dlatego leczenie jest wyłącznie objawowe i polega między innymi na podawaniu leków przeciwdrgawkowych.
Historia
Chorobę opisali jako pierwsi niemieccy neuropatolodzy Hugo Spatz i Julius Hallervorden w pracy z 1922 roku[4][5]. Z powodu zaangażowania tych dwóch lekarzy w nazistowski program przymusowej eutanazji nieuleczalnie chorych dzieci zaproponowano zarzucenie eponimicznej nazwy choroby[6] i zastąpienie jej opisowym określeniem; wyjaśnienie genetycznego podłoża schorzenia pozwoliło na zaproponowanie terminu "neurodegeneracji związanej z kinazą pantotenianową" (pantothenate kinase-associated neurodegeneration)[7].
Przypisy
- ↑ Neurologia wieku rozwojowego. Red. Steinborn, Barbara. Warszawa: PZWL Wydawnictwo Lekarskie, 2017, 1282 s. ISBN 978-83-200-5494-1
- ↑ Angelini L, Nardocci N, Rumi V, Zorzi C, Strada L, Savoiardo M. Hallervorden-Spatz disease: clinical and MRI study of 11 cases diagnosed in life. „Journal of neurology”. 8 (239), s. 417–25, październik 1992. PMID: 1447570.
- ↑ Kumar N, Boes CJ, Babovic-Vuksanovic D, Boeve BF. The "eye-of-the-tiger" sign is not pathognomonic of the PANK2 mutation. „Archives of neurology”. 2 (63), s. 292–3, luty 2006. DOI: 10.1001/archneur.63.2.292. PMID: 16476823.
- ↑ Hallervorden J, Spatz H. Eigenartige Erkrankung im extrapyramidalen System mot besonderer Beteiligung des Globus pallidus und der Substantia nigra: Ein Beitrag zu den Beziehungen zwischen diesen beiden Zentren. „Zeitschrift für die gesamte Neurologie und Psychiatrie”. 79, s. 254-302, 1922.
- ↑ Michael Shevell. Hallervorden and History. „New England Journal of Medicine”. 348. 1, s. 3-4, 2003. PMID: 12510036.
- ↑ J.J. Gordon J.J., Julius Hallervorden, „Neurology”, 43 (7), 1993, s. 1452, PMID: 8327163 .
- ↑ Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KHL, Gitschier J. Genetic, Clinical, and Radiographic Delineation of Hallervorden–Spatz Syndrome. „New England Journal of Medicine”. 348. 33, 2003.
Bibliografia
- Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KHL, Gitschier J. Genetic, Clinical, and Radiographic Delineation of Hallervorden–Spatz Syndrome. „New England Journal of Medicine”. 348. 33, 2003. PMID: 12510040.
- Choroby układu nerwowego. Wojciech Kozubski, Paweł P. Liberski (red.). Warszawa: Wydawnictwo Lekarskie PZWL, 2004, s. 375. ISBN 83-200-2636-9.
Linki zewnętrzne
- NEURODEGENERATION WITH BRAIN IRON ACCUMULATION 1; NBIA1 w bazie Online Mendelian Inheritance in Man (ang.)
- Artykuł z eMedicine (ang.)
- Hallervorden-Spatz syndrome w bazie Who Named It (ang.)
 Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.









