Equação de Butler-Volmer
Em eletroquímica, a equação de Butler-Volmer (em homenagem a John Alfred Valentine Butler[1] e Max Volmer), também conhecida como equação de Erdey-Grúz-Volmer, é uma das relações mais fundamentais da cinética eletroquímica. Ela descreve como a corrente elétrica através de um eletrodo depende da diferença de potencial entre o eletrodo e a porção equipotencial do eletrólito para uma reação redox elementar, simples e rápida, considerando que ocorrem tanto uma reação catódica quanto uma anódica no mesmo eletrodo.[2]
A equação de Butler-Volmer
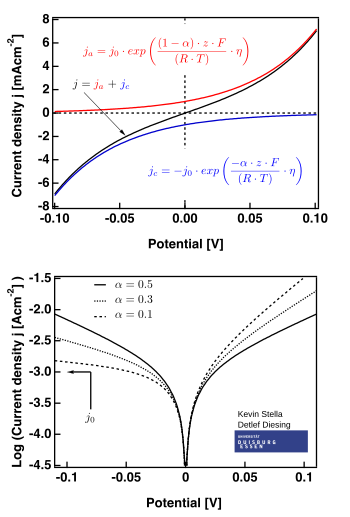
A equação de Butler-Volmer corresponde à expressão abaixo:
![{\displaystyle j=j_{0}\left[e^{{\frac {\alpha _{\rm {a}}zF}{RT}}(E-E_{\rm {eq}})}-e^{-{\frac {\alpha _{\rm {c}}zF}{RT}}(E-E_{\rm {eq}})}\right],}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e8c48c046ef46a83f26811703d58996173d385f0)
ou de uma forma mais compacta,

em que atribuem-se as seguintes denotações:
- : densidade de corrente do eletrodo [L–2 I]
- : densidade de corrente de troca [L–2 I]
- : potencial do eletrodo [T–1 L2 M I–1]
- : potencial de equilíbrio [T–1 L2 M I–1]
- : temperatura absoluta [Θ]
- : número de elétrons envolvidos na reação do eletrodo [1]
- : constante de Faraday [T I N]
- : constante universal dos gases ideais [T–2 L2 M K–1 N–1]
- : coeficiente de transferência de carga catódico [1]
- : coeficiente de transferência de carga anódico [1]
- : sobrepotencial de ativação (definido como ) [T–1 L2 M I–1].












A figura à direita mostra situações nas quais

Os casos limite
Existem dois casos limite da equação de Butler-Volmer:
- a região de baixo sobrepotencial (chamada "resistência de polarização", em que . Nela, a equação de Butler-Volmer pode ser simplificada pela aproximação mostrada abaixo:
- da qual obtém-se a forma linear da equação:



- a região de alto sobrepotencial (em que ), na qual a equação de Butler-Volmer converge para a equação de Tafel. Quando , o primeiro termo dos parênteses domina e, quando , o segundo termo domina.



Além disso, valem as relações:
- para uma reação catódica, quando E<<Eeq, ou
- para uma reação anódica, quando E>>Eeq,


em que e são constantes (para determinadas reação e temperatura) chamadas de constantes da equação de Tafel. Os valores teóricos das constantes da equação de Tafel são diferentes para os processos catódicos e anódicos. No entanto, a inclinação de Tafel pode ser definida como



em que é a corrente faradaica, expressa como , ou seja, a soma das correntes parciais catódica () e anódica ().




A equação de Butler-Volmer estendida
A forma mais generalizada da equação de Butler-Volmer, aplicável às condições influenciadas pela transferência de massa, é apresentada abaixo:[3]
![{\displaystyle j=j_{0}\left[{\frac {c_{\rm {o}}(0,t)}{c_{\rm {o}}^{*}}}\cdot e^{\frac {\alpha _{\rm {a}}zF\eta }{RT}}-{\frac {c_{\rm {r}}(0,t)}{c_{\rm {r}}^{*}}}e^{-{\frac {\alpha _{\rm {c}}zF\eta }{RT}}}\right],}](https://wikimedia.org/api/rest_v1/media/math/render/svg/19a0a96455267cdf76e08687f20d14b6376862f7)
em que se adotam as seguintes denotações:
- : densidade de corrente do eletrodo [L–2 I]
- e são as concentrações, na porção majoritária da solução, das substâncias a serem oxidada e reduzida, respectivamente
- e são as concentrações dependentes do tempo, na porção da solução adjacente ao eletrodo, das substâncias a serem oxidada e reduzida, respectivamente.




A forma acima pode ser simplificada para a convencional (mostrada no início do artigo) quando a concentração da espécie eletroativa próximo à superfície do eletrodo é igual à sua concentração na porção majoritária da solução.
Existem duas taxas que determinam a relação corrente-tensão para um eletrodo. A primeira é a taxa da reação química no eletrodo, que consome reagentes e produz produtos. Isso é conhecido como taxa de transferência de carga. A segunda é a taxa na qual os reagentes são fornecidos e os produtos são removidos da região do eletrodo por vários processos, incluindo difusão, migração e convecção. Esta última é conhecida como taxa de transferência de massa. Essas duas taxas determinam as concentrações dos reagentes e produtos na região do eletrodo, parâmetros que, por sua vez, são determinados pelos próprios eletrodos. A mais lenta dessas taxas determina a taxa geral do processo. Por exemplo, se apenas a difusão contribuir para a taxa de transferência de massa, há uma taxa máxima na qual o reagente pode ser fornecido ao eletrodo e, portanto, a corrente possui um limite superior conhecido como corrente limitante. No caso de processos altamente controlados pela transferência de massa, o valor da densidade de corrente é dado por:

em que:
- é o coeficiente de difusão efetivo (considerando eventuais irregularidades do processo);
- é a espessura da camada de difusão;
- é a concentração da espécie eletroativa limitante na porção majoritária da solução.



A equação de Butler-Volmer simplificada assume que as concentrações no eletrodo são praticamente iguais às concentrações na porção majoritária do eletrólito, de modo que a corrente é expressa apenas em função do potencial. Em outras palavras, assume-se que a taxa de transferência de massa é muito maior que a taxa de transferência de carga e que a reação é dominada pela velocidade da reação química (mais lenta). Apesar dessa limitação, a utilidade da equação de Butler-Volmer na eletroquímica é ampla e muitas vezes é considerada "central na cinética fenomenológica do eletrodo".[4]
A equação estendida de Butler-Volmer não faz essa suposição, mas considera as concentrações no eletrodo como dados conhecidos, produzindo uma relação na qual a corrente é expressa como uma função não apenas do potencial, mas também das concentrações fornecidas. A taxa de transferência de massa pode ser relativamente pequena, mas seu único efeito na reação química é através das diferenças entre as concentrações na porção majoritária da solução e na região próxima ao eletrodo (dados conhecidos). Com efeito, as concentrações também são uma função do potencial. Um tratamento completo, que expressa a corrente apenas em função do potencial, pode ser derivado da equação estendida de Butler-Volmer, mas exige o relacionamento explícito entre os efeitos de transferência de massa e o potencial da solução em cada ponto para que seja possível expressar as concentrações em função unicamente do potencial.
Dedução
Expressão geral
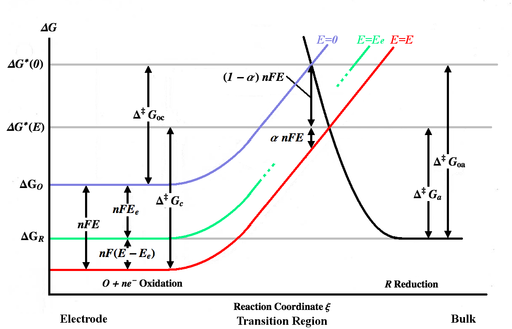
A seguinte derivação da equação estendida de Butler-Volmer é adaptada a partir das deduções de Bard e Faulkner[3] e de Newman e Thomas-Alyea.[5] Para uma reação elementar simples e rápida,

As velocidades das reações direta e inversa ( e ) e as densidades de corrente elétrica associadas a elas ( e ) obtidas a partir das leis de Faraday podem ser escritas como:






em que e são as constantes de velocidade das reações direta e inversa, respectivamente [T–1] e e são as concentrações superficiais das moléculas oxidada e reduzida no eletrodo, respectivamente [N L–2] (denotapas por e na seção anterior). A velocidade líquida () da reação de redução e a densidade de corrente líquida () são, portanto,






Bard[3] define uma corrente proporcional à velocidade líquida de oxidação, mas define o potencial como o potencial do eletrodo menos o potencial do eletrólito, o que tem o desconcertante (mas não inconsistente) efeito de designar uma corrente positiva para um potencial negativo. Na equação acima, é usada a convenção de Newman,[5] na qual a corrente é escolhida proporcionalmente à velocidade líquida de redução, para manter correspondência com os resultados das seções anteriores.
A figura acima representa várias curvas de energia livre de Gibbs em função da coordenada de reação ξ. A coordenada da reação pode ser aproximada para uma medida de distância, com o corpo do eletrodo à esquerda e a solução à direita. A curva azul mostra o aumento na energia livre de Gibbs para uma molécula oxidada conforme ela se aproxima da superfície do eletrodo quando nenhum potencial é aplicado. A curva de energia preta mostra o aumento da energia livre de Gibbs à medida que uma molécula reduzida se aproxima do eletrodo. As duas curvas de energia se cruzam em . A aplicação de um potencial ao eletrodo moverá a curva de energia livre de Gibbs da molécula oxidada para baixo (para a curva vermelha) por e o ponto de interseção se moverá para . Aumentar o potencial dos íons de zero para aumenta seu em , sendo a carga dos íons (ver potencial eletroquímico). Aumentar o potencial do eletrodo diminui o potencial dos íons próximos ao eletrodo em relação a ele, diminuindo, assim, seu . e são as energias de ativação (barreiras energéticas) a serem superadas pelas espécies oxidada e reduzida, respectivamente, para um potencial qualquer, enquanto e são as energias de ativação para . A curva de energia livre de Gibbs da molécula reduzida (curva preta) pode ser afetada pelo potencial, mas as conclusões não são invalidadas desde que a soma dos deslocamentos das curvas de oxidação e redução seja igual a .[5]












Assumindo-se que as constantes de velocidade obedecem à equação de Arrhenius, valem as seguintes relações:


nas quais e são constantes tais que é a frequência de colisões com orientação favorável entre as moléculas oxidada e reduzida e os termos exponenciais (análogos aos fatores de Boltzmann) contabiliza a fração energeticamente favorável dessas colisões.



Supondo-se que as curvas sejam praticamente lineares na região de transição, elas podem ser representadas por:
| (curva azul) | |
| (curva vermelha) | |
| (curva preta) |



O coeficiente de transferência de carga para esse caso simples é equivalente ao fator de simetria e pode ser expresso em termos das inclinações das curvas:

Com isso, obtém-se as seguintes relações:


Por conveniência, define-se:



As constantes de velocidade podem agora ser expressas como:


em que as constantes de velocidade no potencial são:


A densidade de corrente em função do potencial aplicado pode, então, ser escrita como:[5]

Expressão em termos do potencial de equilíbrio
A uma certa tensão , o equilíbrio será atingido e as velocidades das reações direta e inversa ( e ) serão iguais. Isso é representado pela curva verde na figura acima. As constantes de velocidade de equilíbrio serão escritas como e , e as concentrações de equilíbrio serão denotadas por e . As densidades de corrente no equilíbrio ( e ) serão ambas iguais a , a chamada densidade de corrente de troca.










Observe-se que a densidade de corrente líquida no equilíbrio torna-se nula. As constantes de velocidade de equilíbrio são, portanto, dadas por:


Tratando o sistema acima para e em termos das concentrações de equilíbrio e e da densidade de corrente de troca , a densidade de corrente como uma função do potencial aplicado pode ser escrita:[5]


![{\displaystyle j=j_{\mathrm {o} }\left[{\frac {c_{\mathrm {red} }}{c_{\mathrm {red,eq} }}}\,e^{f_{\beta }\,(E-E_{\mathrm {eq} })}-{\frac {c_{\mathrm {ox} }}{c_{\mathrm {ox,eq} }}}\,e^{-f_{\alpha }\,(E-E_{\mathrm {eq} })}\right].}](https://wikimedia.org/api/rest_v1/media/math/render/svg/173ad531c2742f149897540cd5088a05ddd5dfdb)
Assumindo que a porção majoritária da solução se mantém em equilíbrio, com concentrações e , tem-se que e , e a expressão acima para a densidade de corrente se reduz à equação de Butler-Volmer. A diferença é conhecida como sobrepotencial de ativação (denotado por ).





Expressão em termos do potencial formal
Para a reação simplificada mostrada anteriormente, a variação da energia de Gibbs é dada por:

em que e são as atividades no equilíbrio. Note-se que a variação na energia livre de Gibbs também é igual a . As atividades estão relacionadas às concentrações por , sendo o coeficiente de atividade. O potencial de equilíbrio é dado pela equação de Nernst:







em que é o potencial padrão.


Definindo o potencial formal como[3]
o potencial de equilíbrio é, então, dado por:

Substituindo esse potencial de equilíbrio na equação de Butler-Volmer, obtém-se:
![{\displaystyle j={\frac {j_{0}}{c_{\mathrm {ox,eq} }^{1-\alpha }\,c_{\mathrm {red,eq} }^{\alpha }}}\left[c_{\mathrm {red} }\,e^{f_{\beta }(E-E^{o\,\prime })}-c_{\mathrm {ox} }\,e^{-f_{\alpha }(E-E^{o\,\prime })}\right],}](https://wikimedia.org/api/rest_v1/media/math/render/svg/edd6718f0f6ba4d350f2efd3b46cb915c2c16dc4)
que também pode ser escrita em termos da constante de velocidade padrão como:[3]

![{\displaystyle j=n\,F\,k^{o}\left[c_{\mathrm {red} }\,e^{f_{\beta }(E-E^{o\,\prime })}-c_{\mathrm {ox} }\,e^{-f_{\alpha }(E-E^{o\,\prime })}\right].}](https://wikimedia.org/api/rest_v1/media/math/render/svg/de6a994dd29b8025dd04ff98da33cb966717849b)
A constante de velocidade padrão é um importante descritor do comportamento do eletrodo independente das concentrações. É uma medida da velocidade na qual o sistema se aproxima do equilíbrio. No limite, à medida que , o eletrodo se torna um eletrodo polarizável ideal e se comporta eletricamente como um circuito aberto (desconsiderando a capacitância). Para eletrodos quase ideais com pequeno, grandes mudanças no sobrepotencial são necessárias para gerar uma corrente significativa. À medida que , o eletrodo torna-se um eletrodo não-polarizável ideal e se comporta como um sistema em curto-circuito. Para eletrodos quase ideais com altos valores de , pequenas mudanças no sobrepotencial geram grandes mudanças na corrente.


Ligações externas
 Media relacionados com Equação de Butler-Volmer no Wikimedia Commons
Media relacionados com Equação de Butler-Volmer no Wikimedia Commons
Ver também
Referências
- ↑ Mayneord, W. V. (1979). «John Alfred Valentine Butler, 14 February 1899 - 16 July 1977». Biographical Memoirs of Fellows of the Royal Society. 25: 144–178. PMID 11615791. doi:10.1098/rsbm.1979.0004
- ↑ Adler, S.B. (2016). «Chapter 11: Sources of cell and electrode polarisation losses in SOFCs». In: Kendall; Kendall. High-Temperature Solid Oxide Fuel Cells for the 21st Century 2nd ed. [S.l.]: Academic Press. ISBN 9780124104532. doi:10.1016/C2011-0-09278-5
- ↑ a b c d e Bard, Allen; Faulkner, Larry (2001). Electrochemical Methods. Fundamentals and Applications 2ª ed. Hoboken, NJ: John Wiley & Sons, Inc. ISBN 978-0-471-04372-0
- ↑ Reddy, Amulya K.N.; Bockris, John (31 de janeiro de 2001). Modern Electrochemistry 2A. Fundamentals of Electrodics. Nova Iorque: Springer. p. 1083. ISBN 978-0-306-47605-1
- ↑ a b c d e Newman, John; Thomas-Alyea, Karen E. (2004). Electrochemical Systems 3rd ed. Hoboken, NJ: John Wiley & Sons, Inc. ISBN 0-471-47756-7










