Organocatalisi
In chimica organica, con il termine organocatalisi si intende la catalisi di reazioni dove il catalizzatore usato per accelerare la reazione è una piccola molecola organica, priva di elementi inorganici come i metalli e contenente carbonio, idrogeno, zolfo. Il termine fu coniato nel 1935 dal chimico tedesco Wolfgang Langenbeck.[1]
Il 6 ottobre 2021, i chimici David MacMillan e Benjamin List hanno ricevuto il Premio Nobel per la chimica per i loro studi sull'organocatalisi.[2]
Storia

Il primo utilizzo di una molecola organica come catalizzatore è attribuito a Justus von Liebig, che nel 1859 scoprì la trasformazione del cianogeno in ossammide catalizzata da acetaldeide.[3] Liebig identificò l'acetaldeide come catalizzatore della reazione e riconobbe nei suoi effetti un'analogia con i fermenti (enzimi).
La prima reazione di organocatalisi asimmetrica fu pubblicata da Breding e Fiske nel 1912. In questa reazione a partire da benzaldeide si formava una cianidrina usando alcaloidi come catalizzatori. Questi studi furono considerati molto innovativi, anche se l'eccesso enantiomerico raggiunto era minore del 10%.[4]
Decenni dopo fu raggiunta per la prima volta una significativa stereoselettività in una reazione di organocatalisi. Come catalizzatore fu utilizzato l'amminoacido (S) o (R)-prolina in una anellazione di Robinson per ottenere il chetone di Wieland-Miescher. Questa reazione è oggi chiamata reazione di Hajos-Parrish-Eder-Sauer-Wiechert dal nome degli scopritori, ed è di notevole importanza per la sintesi totale di steroidi.[5][6][7]
Con il modello Houk fu per la prima volta proposto un meccanismo coerente, analogo al modello Zimmerman-Traxler, per la reazione aldolica delle enammine senza metalli.[8][9] Reazioni dirette aldoliche incrociate furono sviluppate indipendentemente da List,[10] Barbas,[11] Shibasaki[12] e Trost.[13] La prima reazione organocatalitica aldolica incrociata enantioselettiva di aldeidi è stata sviluppata nel 2002 da MacMillan.[14]
Meccanismo di reazione
Durante il ciclo catalitico il catalizzatore può essere legato covalentemente alla molecola del substrato; in tal caso è necessaria una concentrazione relativamente elevata di catalizzatore organico. Anche attraverso legami non covalenti come i legami idrogeno si possono avere interazioni catalitiche, e in tal caso sono richieste solo piccole quantità di catalizzatore.
Meccanismo covalente
Il principio della maggior parte dei processi organocatalitici è che il catalizzatore viene prima fatto reagire con un reagente per formare (in modo reversibile) un legame covalente. Nella reazione aldolica catalizzata con prolina, la prolina inizialmente dà una reazione di condensazione con il chetone utilizzato. Il risultante catione imminio tautomerizza quindi a enammina, che nel passo successivo dà un attacco nucleofilo sull'aldeide. Per successiva idrolisi viene rilasciato il prodotto e viene riformata la prolina.

Nella reazione, l'informazione stereochimica è determinata dalla prolina chirale. Il gruppo carbossilico della prolina attiva anche l'aldeide mediante formazione di un legame idrogeno. La reazione procede attraverso uno stato di transizione a sei termini con forma a sedia simile al modello di Zimmerman-Traxler per enolati di litio. Il sostituente dell'aldeide sta nel piano pseudo-equatoriale.
Il decorso della reazione attraverso uno stato di transizione a sedia è stato dapprima postulato da Houk in base a calcoli quantomeccanici,[8][9] e poi dimostrato sperimentalmente da List usando ossigeno marcato.[15]
Meccanismo non covalente

In questo meccanismo il catalizzatore non forma legami covalenti. Tra il substrato da attivare e il catalizzatore organico si hanno deboli interazioni direzionali. Questo è il principio con cui reagiscono anche molti enzimi, che vengono utilizzati anche come modello per lo sviluppo di catalizzatori organici non covalenti. In questo campo si utilizzano specie neutre che possono dare legami idrogeno, come i derivati di urea o tiourea.[16][17] Si sono dimostrati buoni catalizzatori quei composti che hanno anelli fenilici di struttura rigida e poveri di elettroni, con sostituenti elettron-attrattori e non coordinanti nelle posizioni 3, 4 o 5.
I vantaggi dei derivati di tiourea (soprattutto rispetto ai catalizzatori tradizionali acidi di Lewis contenenti metalli) sono:
- il catalizzatore si lega al substrato in modo non covalente, il prodotto non dà inibizione
- è sufficiente una piccola quantità di catalizzatore (fino a 0,001 mol%), i valori di turnover frequency sono alti
- la sintesi è semplice e conveniente, con modifiche strutturali
- il catalizzatore può essere legato alla fase solida, rendendone possibile il recupero
- il catalizzatore non è sensibile all'aria o all'acqua, non è necessaria un'atmosfera di gas inerte, non ci sono problemi di manipolazione
- la catalisi è possibile in condizioni quasi neutre, si possono usare substrati labili in presenza di acidi
- il catalizzatore non contiene metalli e non è tossico, a differenza di molti catalizzatori acidi di Lewis contenenti metalli
- il catalizzatore è più ecologico ("chimica verde")
Reazioni
Alcuni tipi di reazione che si possono condurre efficacemente tramite organocatalisi sono:
- Reazione aldolica
- Condensazione di Knoevenagel
- Reazione asimmetrica di Diels-Alder
- Reazione asimmetrica di Michael
- Reazione asimmetrica di Mannich
- Epossidazione di Shi
- Reazione di Stetter
- Reazione di Baylis-Hillman
Catalizzatori derivati da prodotti naturali
I derivati dell'amminoacido (S)-prolina sono stati e sono tuttora comunemente utilizzati.[18][19] Sono spesso usati anche derivati della (S)-fenilalanina:

Altri catalizzatori sono derivati di alcaloidi contenuti nella Cinchona:

Si utilizzano anche derivati dell'acido tartarico, come ad esempio il TADDOL:

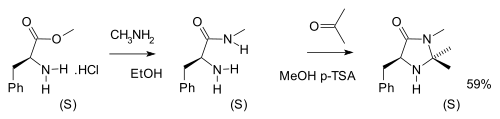
Dalla biomolecola fenilalanina si ottengono i catalizzatori di MacMillan, usati in molte reazioni di catalisi asimmetrica. La sintesi in due passaggi lascia intatta la chiralità:[20]
Note
- ^ Langenbeck 1935
- ^ 2021 Nobel Prize in chemistry, su Nobel Prize, Nobel Prize. URL consultato il 6 ottobre 2021.
- ^ von Liebig 1860
- ^ Bredig e Fiske 1912
- ^ Eder et al. 1971
- ^ Hajos e Parrish 1971
- ^ Hajos e Parrish 1974
- ^ a b Houk e Bahmanyar 2001
- ^ a b Bahmanyar e Houk 2001
- ^ List et al. 2000
- ^ Kandasamy et al. 2001
- ^ Yamada et al. 1997
- ^ Trost e Ito 2000
- ^ Northrup e MacMillan 2002
- ^ Hoang et al. 2003
- ^ Taylor e Jacobsen 2006
- ^ Connon 2006
- ^ Seebach et al. 2007
- ^ Mukherjee et al. 2007
- ^ Ahrendt et al. 2000
Bibliografia
- K. A. Ahrendt, C. J. Borths e D. W. C. MacMillan, New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels-Alder Reaction, in J. Am. Chem. Soc., vol. 122, n. 17, 2000, pp. 4243–4244, DOI:10.1021/ja000092s. URL consultato il 5 aprile 2014.
- S. Bahmanyar e K. N. Houk, Transition States of Amine-Catalyzed Aldol Reactions Involving Enamine Intermediates: Theoretical Studies of Mechanism, Reactivity, and Stereoselectivity, in J. Am. Chem. Soc., vol. 123, n. 45, 2001, pp. 11273-11283, DOI:10.1021/ja011403h. URL consultato il 3 aprile 2014.
- G. Bredig e P. S. Fiske, Durch Katalysatoren Bewirkte Asymmetrische Synthese, in Biochem. Z., vol. 46, n. 7-23, 1912.
- S. J. Connon, Organocatalysis Mediated by (Thio)urea Derivatives, in Chem. Eur. J., vol. 12, n. 21, 2006, pp. 5418 –5427, DOI:10.1002/chem.200501076. URL consultato il 3 aprile 2014.
- U. Eder, G. Sauer e R. Wiechert, Neuartige asymmetrische Cyclisierung zu optisch aktiven Steroid-CD-Teilstücken, in Angew. Chem., vol. 10, n. 13, 1971, pp. 492–493, DOI:10.1002/ange.19710831307. URL consultato il 3 aprile 2014.
- Z. G. Hajos e D. R. Parrish, Asymmetric Synthesis of Optically Active Polycyclic Organic Compounds, in German Patent DE 2102623, 29 luglio 1971.
- Z. G. Hajos e D. R. Parrish, Asymmetric synthesis of bicyclic intermediates of natural product chemistry, in J. Org. Chem., vol. 39, n. 12, 1974, pp. 1615-1621, DOI:10.1021/jo00925a003. URL consultato il 3 aprile 2014.
- L. Hoang, S. Bahmanyar, K. N. Houk e B. List, Kinetic and Stereochemical Evidence for the Involvement of Only One Proline Molecule in the Transition States of Proline-Catalyzed Intra- and Intermolecular Aldol Reactions, in J. Am. Chem. Soc., vol. 125, n. 1, 2003, pp. 16-17, DOI:10.1021/ja028634o. URL consultato il 3 aprile 2014.
- K. N. Houk, S. Bahmanyar, The Origin of Stereoselectivity in Proline-Catalyzed Intramolecular Aldol Reactions, in J. Am. Chem. Soc., vol. 123, n. 51, 2001, pp. 12911-12912, DOI:10.1021/ja011714s. URL consultato il 3 aprile 2014.
- S. Kandasamy, W. Notz, T. Bui e C. F. Barbas, III., Amino Acid Catalyzed Direct Asymmetric Aldol Reactions: A Bioorganic Approach to Catalytic Asymmetric Carbon−Carbon Bond-Forming Reactions, in J. Am. Chem. Soc., vol. 123, n. 22, 2001, pp. 5260–5267, DOI:10.1021/ja010037z. URL consultato il 3 aprile 2014.
- (DE) W. Langenbeck, Die organischen Katalysatoren und ihre Beziehungen zu den Fermenten, Berlin, Springer, 1935.
- B. List, R. A. Lerner e C. F. Barbas, III, Proline-Catalyzed Direct Asymmetric Aldol Reactions, in J. Am. Chem. Soc., vol. 122, n. 10, 2000, pp. 2395–2396, DOI:10.1021/ja994280y. URL consultato il 3 aprile 2014.
- S. Mukherjee, J. W. Yang, S. Hoffmann e B. List, Asymmetric enamine catalysis, in Chem. Rev., vol. 107, n. 12, 2007, pp. 5471–5569, DOI:10.1021/cr0684016. URL consultato il 5 aprile 2014.
- A. B. Northrup e D. W. C. MacMillan, The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes, in J. Am. Chem. Soc., vol. 124, n. 24, 2002, pp. 6798-6799, DOI:10.1021/ja0262378. URL consultato il 3 aprile 2014.
- D. Seebach, A. K. Beck , D. M. Badine, M. Limbach, A. Eschenmoser, A. M. Treasurywala, R. Hobi, W. Prikoszovich, B. Linder, Are Oxazolidinones really unproductive, parasitic species in proline catalysis? Thoughts and experiments pointing to an alternative view, in Helv. Chim. Acta, vol. 90, n. 3, 2007, pp. 425–471, DOI:10.1002/hlca.200790050. URL consultato il 5 aprile 2014.
- M. S. Taylor e E. N. Jacobsen, Asymmetric Catalysis by Chiral Hydrogen-Bond Donors, in Angew. Chem. Int. Ed., vol. 45, n. 10, 2006, pp. 1520–1543, DOI:10.1002/anie.200503132. URL consultato il 3 aprile 2014.
- B. M. Trost e H. Ito, A Direct Catalytic Enantioselective Aldol Reaction via a Novel Catalyst Design, in J. Am. Chem. Soc., vol. 122, n. 48, 2000, pp. 12003–12004, DOI:10.1021/ja003033n. URL consultato il 3 aprile 2014.
- J. von Liebig, Ueber die Bildung des Oxamids aus Cyan, in Justus Liebigs Ann. Chem., vol. 113, n. 2, 1860, pp. 246–247, DOI:10.1002/jlac.18601130213. URL consultato il 3 aprile 2014.
- Y. M. A. Yamada, N. Yoshikawa, H. Sasai e M. Shibasaki, Direkte katalytische asymmetrische Aldolreaktionen von Aldehyden mit nicht modifizierten Ketonen, in Angew. Chem., vol. 109, n. 17, 1997, pp. 1842–1944, DOI:10.1002/ange.19971091716. URL consultato il 3 aprile 2014.
Altri progetti
Altri progetti
- Wikimedia Commons
 Wikimedia Commons contiene immagini o altri file su Organocatalisi
Wikimedia Commons contiene immagini o altri file su Organocatalisi
V · D · M | ||
|---|---|---|
| Isomeria | Stereoisomero · Enantiomero · Diastereoisomero · Epimero · Composto meso |  |
| Chiralità | Elemento stereogenico · Chiralità planare · Ligando chirale · Chiralità assiale · Chiralità inerente · Prochiralità · Omochiralità · Racemo · Anomeria · | |
| Sintesi | Stereospecificità · Stereoselettività · Eccesso enantiomerico · Eccesso diastereoisomerico | |
| Analisi | Potere rotatorio · Risoluzione chirale · Dicroismo circolare | |
| Reazioni | Sintesi asimmetrica · Ausiliario chirale · Organocatalisi · Biocatalisi | |
 Portale Chimica: il portale della scienza della composizione, delle proprietà e delle trasformazioni della materia
Portale Chimica: il portale della scienza della composizione, delle proprietà e delle trasformazioni della materia








